

Discover the Oncodesign Services Computer Aided Drug Design (CADD) in detail
Why use a computer aided drug design in your drug discovery programs?
Computer-Aided Drug Design (CADD) has been used for many years in the pharmaceutical research sector. The CADD provides support for experiments throughout the research process of drug candidate, from the identification of biological targets to the first pre-clinical studies.
The primary objective of CADD is to accelerate the research process by helping medicinal chemists to guide the strategic choices of drug candidates.
Support your Drug Discovery process with CADD
SBBD and LBDD : 2 complementary methods
Two major approaches are conventionally used in CADD: the ligand-based drug design (LBDD) which is mainly based on data associated with small molecules in the absence of 3D structure of the target of interest, and the structure-based drug design (SBDD) which uses the 3D structure of a biological target to help in the selection and sorting of drug candidates.
Several methodologies associated with LBDD or SBDD can be used with Computer-Aided Drug Design process :
Methods of SBDD :
- Binding site search and binding mode analysis
- Pharmacophore-based docking
- Covalent docking
- Virtual screening
Methods of LBDD :
- Pharmacophore modelling
- QSAR-analysis
- R-group decomposition
- Free-Wilson analysis
- Activity prediction
Oncodesign Services offers computational chemistry services based on CADD experimented team.
In close collaboration the with the in vitro, in vivo , DMPK and bioanalytical departments, the medicinal chemistry unit works to deliver quality hits, leads and drug candidates with fast cycles.
The CADD team can carry out the following tasks:
1
Target analysis and pocket finding
Starting from a 3D protein structure, we can help you finding the actual binding site and the way the ligand binds the protein. If no X-ray exists, we use the power of AI and AlphaFold predictions to find a potential binding site.
2
Docking and binding mode analysis
Based on 6 different scores implemented into our MOE software, we can easily identify if your ligand anchors into the binding site and the way it binds. We can use a free approach, a pharmacophore-based approach or a covalent docking approach depending on your own problematic.
3
Virtual screening
Based on shape, pharmacophores or predicted properties (phys-chem properties, toxicity, synthesis accessibility scoring…) and activities, we can help you to quickly screen chemical libraries (private and commercial) for hit finding. We are also able to generate in silico chemicals by library combination.
4
SAR analysis and lead optimization
To help your chemistry team, we can analyze your own data and exploit other libraries with chemicals space analysis and R-group analysis for instance. We can also suggest you some modifications with scaffold replacement and Free Wilson suggestions.
Starting from a 3D protein structure, we can help you finding the actual binding site and the way the ligand binds the protein. If no X-ray exists, we use the power of AI and AlphaFold predictions to find a potential binding site.
Based on 6 different scores implemented into our MOE software, we can easily identify if your ligand anchors into the binding site and the way it binds. We can use a free approach, a pharmacophore-based approach or a covalent docking approach depending on your own problematic.
Based on shape, pharmacophores or predicted properties (phys-chem properties, toxicity, synthesis accessibility scoring…) and activities, we can help you to quickly screen chemical libraries (private and commercial) for hit finding. We are also able to generate in silico chemicals by library combination.
To help your chemistry team, we can analyze your own data and exploit other libraries with chemicals space analysis and R-group analysis for instance. We can also suggest you some modifications with scaffold replacement and Free Wilson suggestions.
Oncodesign Services uses a combination of software in CADD. The MOE (Molecular Operating Environment) software has been in place at Oncodesign Services for many years, and combines efficiently SBDD and LBDD approaches. This software is completed by KNIME, which offers automatize and speed up computational workflows for LBDD and SBDD.
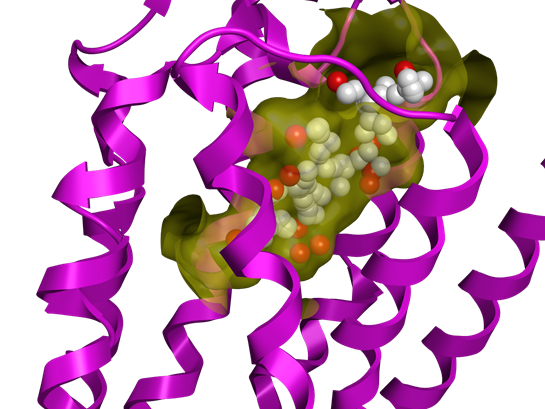
AlphaShapes methodology is used to search for the potential binding site. Dummy atoms (red and white) represents it and are well placed into the receptor surface (in yellow)
Case study : Use of AlphaFold2 to discover potential binding mode
Aim of the study:
- Help our client to understand the mode of action of their lead
- Without co-crystalized structure available
- Without any idea of the binding mode
Materials:
- 3 active ligand structures (same series)
- AlphaFold2 (AF2) protein structure prediction (GPCR)
- MOE software from CCG (Chemical Computing Group)
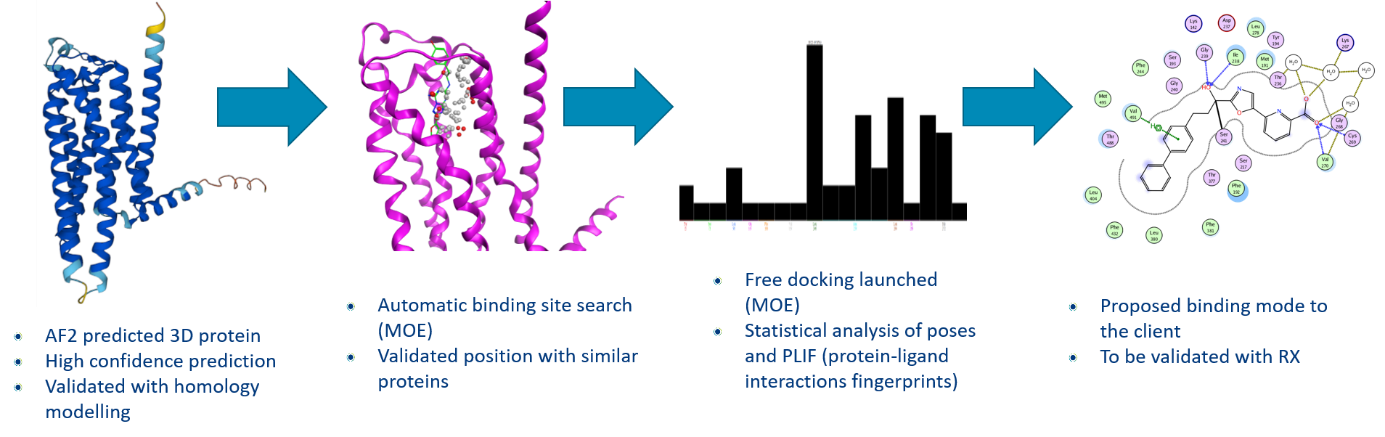
Conclusion of the case study : binding mode proposals validated by experimental in vitro data.

