Découvrez nos services de conception de médicaments assistée par ordinateur (CADD)
Pourquoi utiliser la conception de médicaments assistée par ordinateur pour vos programmes de drug discovery ?
La conception de médicaments assistée par ordinateur (Computer-Aided Drug Design ou CADD en anglais) est utilisée depuis de nombreuses années dans le secteur de la recherche pharmaceutique. La CADD apporte un soutien aux tests tout au long du processus de recherche d’un candidat-médicament, depuis l’identification des cibles biologiques jusqu’aux premières études précliniques.
L’objectif premier de la CADD est d’accélérer le processus de recherche en aidant les chimistes médicinaux à orienter les choix stratégiques des candidats-médicaments.
Accélérer vos programmes de drug discovery avec la CADD
SBBD et LBDD : 2 méthodes complémentaires
Deux approches majeures sont classiquement utilisées dans la conception de médicament assistée par ordinateur : la conception de médicaments basée sur les ligands (LBDD) qui est principalement basée sur les données associées aux petites molécules en l’absence de structure 3D de la cible d’intérêt, et la conception de médicaments basée sur la structure (SBDD) qui utilise la structure 3D d’une cible biologique pour aider à la sélection et au tri des candidats-médicaments.
Plusieurs méthodologies associées à la LBDD ou à la SBDD peuvent être utilisées dans le cadre du processus de conception de médicaments assistée par ordinateur :
Méthodes de SBDD :
- Recherche de sites de liaison et analyse des modes de liaison
- Arrimage basé sur les pharmacophores
- Arrimage covalent
- Criblage virtuel
Méthodes de LBDD :
- Modélisation pharmacophore
- Analyse QSAR
- Décomposition en groupes R
- Analyse de Free-Wilson
- Prédiction d’activité
Oncodesign Services offre des services sur-mesure de conception de médicament assistées par ordinateur
En étroite collaboration avec les départements in vitro, in vivo, DMPK et bioanalytique, l’unité de chimie médicinale s’efforce de fournir des hits, des leads et des candidats-médicaments de qualité avec des cycles rapides.
L’équipe CADD peut effectuer les tâches suivantes :
1
Analyse des cibles et recherche de poches
À partir d’une structure de protéine en 3D, nous pouvons vous aider à trouver le site de liaison réel et la manière dont le ligand se lie à la protéine. Si aucune radiographie n’existe, nous utilisons la puissance de l’IA et des prédictions AlphaFold pour trouver un site de liaison potentiel.
2
Docking et analyse du mode de liaison
Sur la base de 6 scores différents implémentés dans notre logiciel MOE, nous pouvons facilement identifier si votre ligand s’ancre dans le site de liaison et la manière dont il se lie. Nous pouvons utiliser une approche libre, une approche basée sur le pharmacophore ou une approche d’amarrage covalent en fonction de votre propre problématique.
3
Criblage virtuel
Sur la base de la forme, des pharmacophores ou des propriétés prédites (propriétés physico-chimiques, toxicité, accessibilité à la synthèse, notation…) et des activités, nous pouvons vous aider à cribler rapidement des chimiothèques (privées et commerciales) pour trouver des résultats. Nous sommes également en mesure de générer des produits chimiques in silico par combinaison de bibliothèques.
4
Analyse SAR et optimisation des pistes
Pour aider votre équipe de chimie, nous pouvons analyser vos propres données et exploiter d’autres bibliothèques avec l’analyse de l’espace chimique et l’analyse des groupes R par exemple. Nous pouvons également vous suggérer des modifications avec le remplacement d’échafaudages et les suggestions de Free Wilson.
À partir d’une structure de protéine en 3D, nous pouvons vous aider à trouver le site de liaison réel et la manière dont le ligand se lie à la protéine. Si aucune radiographie n’existe, nous utilisons la puissance de l’IA et des prédictions AlphaFold pour trouver un site de liaison potentiel.
Sur la base de 6 scores différents implémentés dans notre logiciel MOE, nous pouvons facilement identifier si votre ligand s’ancre dans le site de liaison et la manière dont il se lie. Nous pouvons utiliser une approche libre, une approche basée sur le pharmacophore ou une approche d’amarrage covalent en fonction de votre propre problématique.
Sur la base de la forme, des pharmacophores ou des propriétés prédites (propriétés physico-chimiques, toxicité, accessibilité à la synthèse, notation…) et des activités, nous pouvons vous aider à cribler rapidement des chimiothèques (privées et commerciales) pour trouver des résultats. Nous sommes également en mesure de générer des produits chimiques in silico par combinaison de bibliothèques.
Pour aider votre équipe de chimie, nous pouvons analyser vos propres données et exploiter d’autres bibliothèques avec l’analyse de l’espace chimique et l’analyse des groupes R par exemple. Nous pouvons également vous suggérer des modifications avec le remplacement d’échafaudages et les suggestions de Free Wilson.
Oncodesign Services utilise une combinaison de logiciels en CDAO. Le logiciel MOE (Molecular Operating Environment) est en place chez Oncodesign Services depuis de nombreuses années et combine efficacement les approches SBDD et LBDD. Ce logiciel est complété par KNIME, qui permet d’automatiser et d’accélérer les processus de calcul pour les approches LBDD et SBDD.
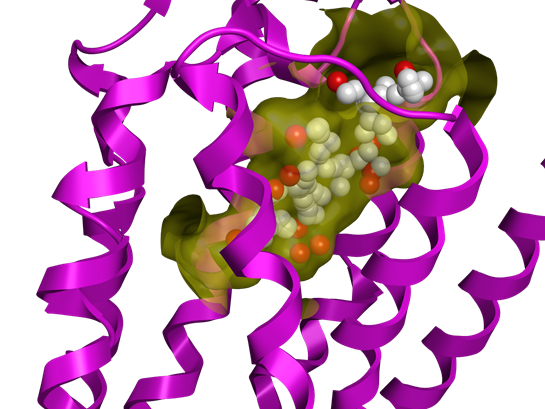
La méthodologie AlphaShapes est utilisée pour rechercher le site de liaison potentiel. Les atomes fictifs (rouge et blanc) le représentent et sont bien placés dans la surface du récepteur (en jaune).
Étude de cas : Utilisation d’AlphaFold2 pour découvrir un mode de liaison potentiel
Objectif de l’étude :
- Aider notre client à comprendre le mode d’action de son plomb.
- Sans structure co-cristallisée disponible
- Sans aucune idée du mode de liaison
Matériaux :
- 3 structures de ligands actifs (même série)
- Prédiction de la structure de la protéine AlphaFold2 (AF2) (GPCR)
- Logiciel MOE du GCC (Groupe de calcul chimique)
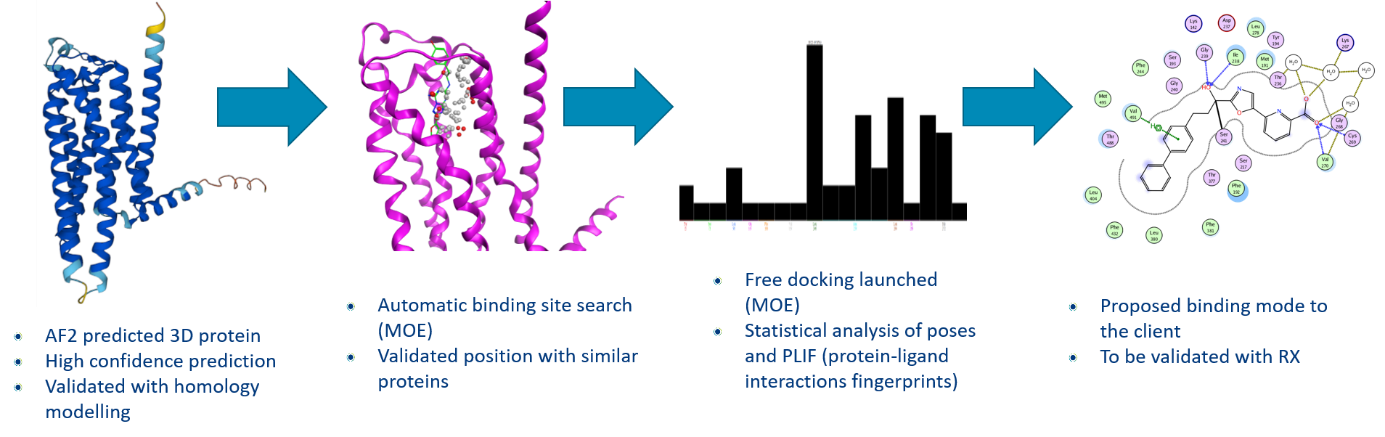
Conclusion de l’étude de cas : les propositions de mode de liaison sont validées par des données expérimentales in vitro.